 |
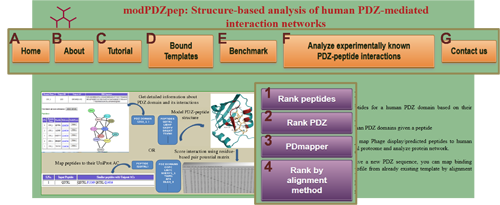
Home page provides four ways to work on PDZ-domain network prediction. |
modPDZpep: Strucure-based analysis of human PDZ-mediated interaction networks |
| Home | About | Tutorial | Bound Templates | Benchmark | Analyze experimentally known PDZ-peptide interactions | Contact us |
A. "Home" Home page of modPDZpep
|
Home page provides four ways to work on PDZ-domain network prediction. |
A1. Rank peptides


|
By clicking on the "Rank Peptides", you will be redirected to a input page where you have
to select a query PDZ domain and enter the single/multiple peptide sequence(s). You can also provide Uniprot accession numbers and length of
peptide to retrieve the protein C-terminal sequences automatically or you can upload a file having peptide sequence(s) or Uniprot AC(s) by
clicking on the Browse button. If length of peptide is not provided with Uniprot AC, then default length of five will be used. Here it should
be noted that the length of peptide for modeling in binding pocket of PDZ is restricted to the number of residues as present in the template
PDZ (in PDB) used for modeling that structure. |
A2. Rank PDZ
 |
With this input method, you can select multiple PDZ domains by pressing (Ctrl+click)) or (Shift+click) to fetch the score of a peptide with all of them. Again here, peptide sequence or Uniprot AC with length of peptide can be entered. The result page provides the PDZ-score table having PDZ domain name linked to SWISS-MODEL modeling report and PDB ID to PDB web page. The binding pocket residues and structural model of the highest ranked PDZ-peptide interaction is also shown on this page. Any of the PDZ-peptide model can be visulaized by selecting it and clicking on the 'Get Selected Model'. |
A3. PDmapper
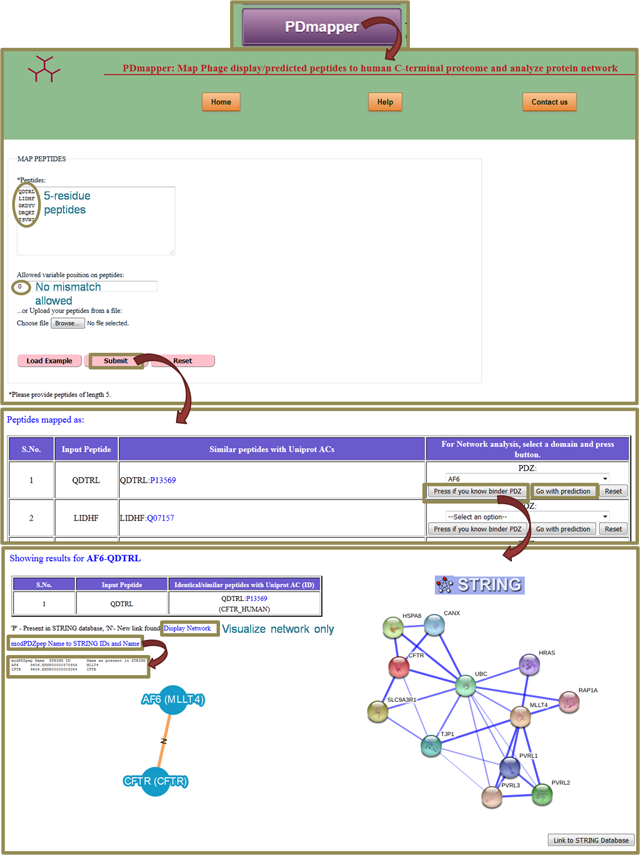 |
PDmapper is a tool which facilitates the mapping of phage display/predicted peptides to Uniprot AC, thus helps in filtering of human proteomic peptides from random peptides. It also tries to find out similar peptides in C-terminal proteome and generate a protein-protein network. For more details, go to PDmapper Help page. |
A4. Rank by alignment method (mapping binding pocket residues from already existing template PDZ)
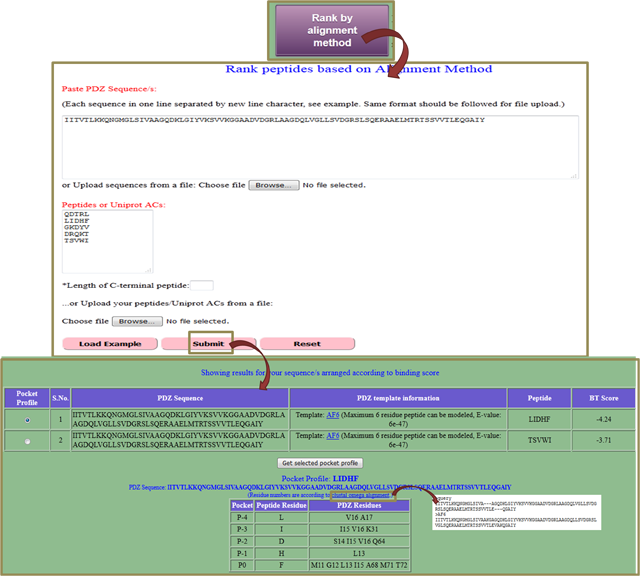 |
If you do not find your query PDZ domain in PDZ domains listed by us or you have a new PDZ sequence, then you can go for this link. It based on the sequence similarity of query PDZ sequence to already saved PDZ sequences, finds a template PDZ-peptide structure to model binding pocket residues and then get BT-score for peptide. You can input multiple PDZs or peptides separated by new line to it (if you are providing Uniprot AC, then do not forget to mention length of peptide to be used). After submitting the job, you get a pairwise list of BT score for PDZs and peptides where domain name is linked to SWISS MODEL report page of PDZ domain. Also, the binding pocket profile for highest ranked interaction is shown but you can get binding pocket residues for any interaction by selecting that interaction and clicking on 'Get selected pocket profile'. For visualizing the alignment between query PDZ and template PDZ sequence, click on clustal omega alignment. |
B. "About" General information on modPDZpep workflow
C. "Tutorial" This Page
D. "Bound Templates" List of peptide bound templates used for modeling PDZ-peptide structures
E. "Benchmark" Benchmarking results of modPDZpep structure-based approach
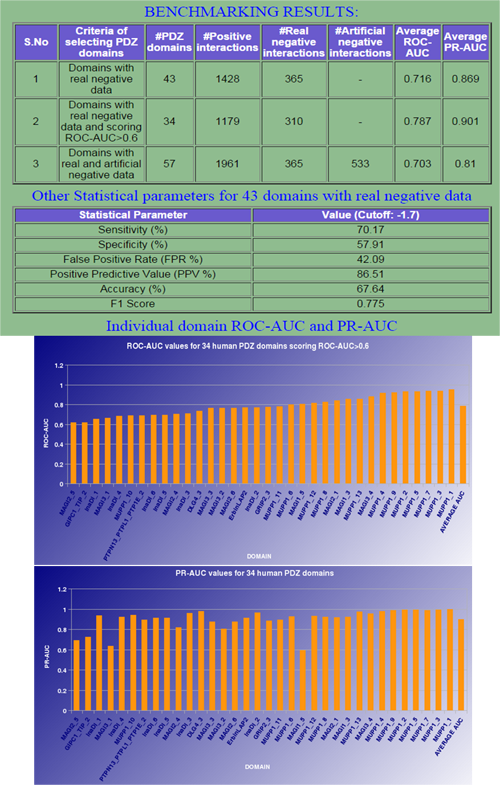 |
It provides the detailed information on benchamrking dataset with number of PDZ domains, binder and non-binder peptides used and their ROC/PR-AUC values with other statistical parameters like Sensitivity, Specificity, FPR, PPV, Accuracy and F1 Score. Individual domains performance can also be visualized on this page. |
F. "Analyze experimentally known PDZ-peptide interactions" Link to experimental data cataloged in modPDZpep
I. "Contact us" Contact information
back to top